
El valor más grande de un profesional es su potencial de hacer la diferencia y usarlo para sumar en su ámbito de acción Mónica
Velasco y Lourdes Molina, 16 ENE 2024
Es altamente recomendable que los medicamentos y sus procesos de fabricación sean diseñados y desarrollados conforme a los principios de “Quality by Design” (QbD) referido en la ICH Q8, cuyos elementos principales son los siguientes:
- El perfil de calidad objetivo del producto (QTPP) que identifica los atributos críticos de calidad (CQAs),
- Diseño del producto incluyendo la identificación de los atributos críticos de los materiales (CMAs)
- Diseño del proceso e identificación de los parámetros críticos del proceso (CPPs)
- La vinculación de los CMAs y CPPs con los CQAs
- La estrategia de control que incluye las especificaciones del producto, materiales y excipientes, así como los controles para cada etapa del proceso de fabricación
- La capacidad del proceso y la mejora continua.
El conocimiento obtenido en la fase de diseño y desarrollo del proceso en conjunto con las herramientas de QbD como la evaluación de riesgos, modelos, diseño de experimentos, análisis de datos y la tecnología analítica de proceso (PAT) aplicados para obtener los datos de la fabricación de los lotes piloto, la transferencia de tecnología y el escalamiento de lotes a tamaño comercial, completan el ciclo de QbD Dicha información, sirve de punto de partida para construir el protocolo de validación del proceso y subsecuentemente, el programa de verificación continua de proceso (ICH Q10). Las tres etapas de validación de procesos van a depender de estudios de gestión de riesgo (ICH Q 9), a fin de mantener el proceso de fabricación del producto en estado de control durante todo el ciclo de vida de este. Cabe mencionar que los estudios de estabilidad que derivan tanto de la fabricación de los lotes piloto como de los lotes de validación generan datos muy valiosos con relación al comportamiento del producto en las condiciones de almacenamiento durante su vida de anaquel. Para algunas agencias regulatorias la validación de los lotes piloto antecede a la transferencia de tecnología y al escalamiento. Sin embargo, existen consideraciones regulatorias en las que se requiere la validación de 1 o 2 lotes a escala comercial (EMA)
Los procesos y los productos pueden sufrir cambios o mejoras (ICH Q10) a lo largo de su ciclo de vida y mientras más conocimiento se tenga sobre la etapa de diseño, será más fácil verificar el espacio de diseño, es decir, los rangos normales de operación que puede manejar el proceso, lo que permite determinar con eficacia el impacto en el Estado Validado de este y por ende en la calidad del producto. Cuando los cambios no exceden el espacio de diseño, se debe documentar el cambio en un control de cambios, implementarlo y reportarlo en el Revisión Anual de Producto (ICH Q 10). Pero si el cambio excede el espacio de diseño, es decir los rangos normales de operación, entonces es necesario documentar el cambio (ICH Q 10), revalidar parcial o totalmente el proceso y cuando aplique solicitar autorización a la agencia regulatoria. Estos procesos involucran la generación y análisis de una gran cantidad de datos cuyas conclusiones están plasmadas en el CMC (Chemical, Manufacturing and Control). Por lo tanto, el CMC que corresponde con el capítulo 3 del CTD debe ser actualizado (ICH Q12).
Una consideración muy importante es que la Validación del proceso debe realizarse en forma prospectiva con la opción de liberación concurrente de los lotes de validación, siendo esta última permitida cuando los productos son de demanda limitada, tienen vidas medias cortas o por emergencia sanitaria.
El Mantenimiento del Estado Validado se puede lograr a través de la Verificación Continua del Proceso, ya que esta involucra una evaluación de tendencias en la que se pueden observar los cambios en la variabilidad y la forma en que se volvió a llevar el proceso a su estado de control mediante, la adecuada gestión de las desviaciones o no conformidades (ICH Q 10). La variabilidad también se puede detectar mediante la evaluación de:
- Quejas relacionadas al producto
- Variaciones en el rendimiento
- Anomalías encontradas durante la revisión de expedientes de lotes
- Registros de recepción de insumos
- Reportes de eventos adversos.
- Eventos de mantenimiento correctivo
- Eventos de calibración
- Entre otros.
El mantenimiento del estado validado debe documentarse apropiadamente y debe incluir una conclusión (ICH Q10):
- El proceso mantuvo su estado validado
- El proceso requiere revalidación parcial
- El proceso requiere revalidación total
es su potencial de hacer la diferencia y usarlo para sumar en su ámbito de acción
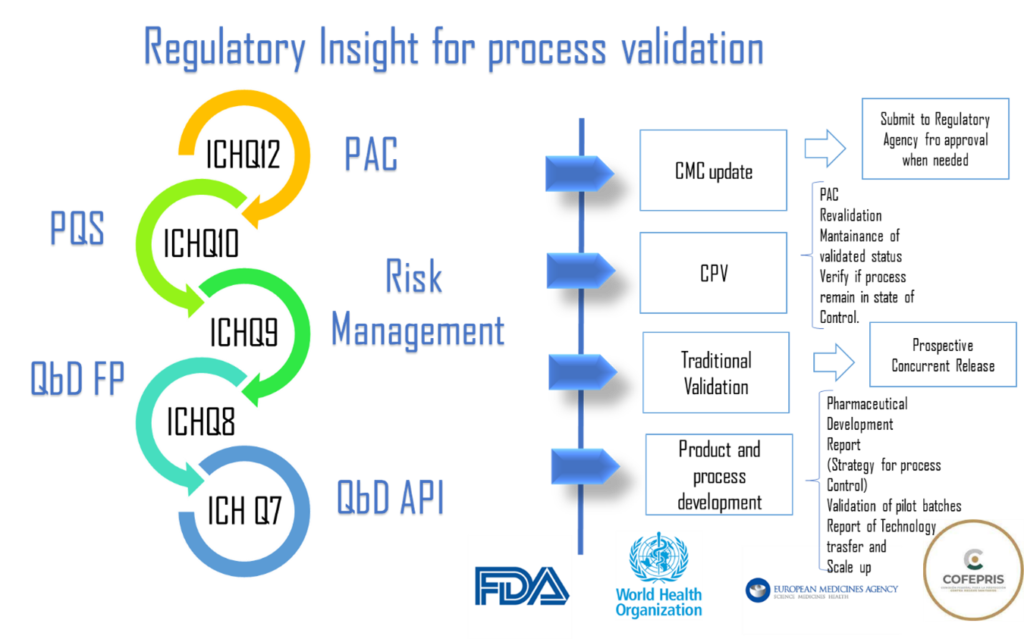
La Figura 1 es un mapa mental que representa en forma general todos los requerimientos para lograr una validación de proceso y una verificación continua sólida basada en ciencia y en cumplimiento de los requerimientos regulatorios aplicables a FDA, EMA y OMS.
La fórmula y el proceso de fabricación de un producto desarrollado por una empresa local, son puntos clave a revisar durante la evaluación de una solicitud de Registro Sanitario. Los funcionarios de la Agencia Regulatoria, basan su evaluación en los siguientes documentos:
- Informe de desarrollo farmacéutico que incluye la estrategia de control, obtenido de los datos generados durante la fabricación de lotes piloto y estudios de estabilidad acelerada (3M) y largo plazo (6M).
- Órdenes de fabricación de los lotes piloto con los que se corrieron los estudios de estabilidad.
- Protocolos de transferencia y escalamiento.
- Especificaciones de materias primas, materiales y producto.
- Protocolos de validación de proceso.
- Métodos de análisis de materias primas, materiales y producto.
- Reporte de validación de los métodos analíticos de materias primas, materiales y producto, cuando no sean farmacopéicos.
- Certificado de análisis de materias primas y materiales usados en la fabricación de los lotes piloto con los que se hicieron los estudios de estabilidad, emitido por el fabricante.
- Certificados analíticos de los lotes piloto.
- Proyectos de marbete.
- Resultados de los estudios de intercambiabilidad o Conclusiones de los estudios clínicos, según aplique.
- Plan de Manejo de Riesgo de Farmacovigilancia.
- Certificado de Cumplimiento de Buenas Prácticas de Fabricación del establecimiento fabricante del principio activo y del establecimiento fabricante del producto terminado.
Por otro lado, la fórmula y el proceso de fabricación de un producto de importación, también se revisan con profundidad durante la evaluación de una solicitud de Registro Sanitario. En este caso los funcionarios basan su evaluación en los siguientes documentos:
- Informe de desarrollo farmacéutico.
- Reportes de transferencia y escalamiento.
- Especificaciones de materias primas, materiales y producto.
- Reporte de validación de proceso.
- Reportes de validación de métodos analíticos de materias primas, materiales y producto terminado, cuando no sean farmacopéicos.
- Órdenes de fabricación de los lotes de validación con los que se hicieron los estudios de estabilidad.
- Estudios de estabilidad acelerada (3M) y largo plazo v (6M) de los lotes de validación con los que se corrieron los estudios de estabilidad.
- Certificado de análisis de materias primas y materiales utilizados en la fabricación de los lotes de validación con los que se hicieron los estudios de estabilidad, emitido por el fabricante.
- Certificados de análisis de los lotes de validación.
- Proyectos de marbete.
- Resultados de los estudios de intercambiabilidad o Conclusiones de los estudios clínicos.
- Reporte Periódico de Seguridad.
- Certificado de Cumplimiento de Buenas Prácticas de Fabricación del establecimiento fabricante del principio activo y del establecimiento fabricante del producto terminado.
Subsecuentemente, ellos cruzan todos los datos para verificar que el proceso y su estrategia de control están alineados con las especificaciones que debe cumplir el producto y las condiciones de almacenamiento establecidas en el proyecto de marbete. Los estudios de estabilidad tienen el propósito para verificar que el sistema contenedor cierre y la fórmula son capaces de mantener su integridad, a lo largo de la vida de anaquel.
La figura 2 muestra el fundamento normativo de la revisión de la fórmula y el proceso que hacen los funcionarios de la Agencia Regulatoria cuando evalúan la solicitud del Registro Sanitario de un medicamento.
Figura 2.

Con lo dicho anteriormente, es posible concluir que la calidad con que se haga el desarrollo del producto y el diseño del proceso es preponderante para desarrollar estudios de validación y de verificación continua que ofrezcan procesos en estado de control con los que se fabriquen productos que cumplan consistentemente con los atributos de calidad, seguridad y eficacia establecidos. También podemos concluir que el registro sanitario depende de la robustez de la estrategia de control y de su alineación con los atributos de calidad del producto, dado un espacio de diseño bien caracterizado. Finalmente, el mantenimiento del CBPF y el mantenimiento del Registro Sanitario dependen de la gestión de los procesos de fabricación mediante la gestión eficaz del Sistema de Calidad Farmacéutica (ICH Q10) y el Sistema de Gestión de Riesgos (ICH Q 9).
Si quieres saber más del tema, pregunta por nuestro curso cerrado de validación de procesos y nuestros servicios de asesoría al respecto, contáctanos en contacto@pharmaclims.com y también puedes llamar a Lourdes Molina al 5528649003 o a Jesús Quintana al 5539892647. También te invito a suscribirte a La Gaceta del Concejal que es nuestra Newsletter quincenal. Finalmente, te invitamos a visitar el perfil de Pharma CLIM Services en LinkedIn y nuestra página web en www.pharmaclims.com.