
El valor más grande de un profesional es su potencial de hacer la diferencia y usarlo para sumar en su ámbito de acción.
Autora: Lourdes Amelia Molina Rincón, 19 de diciembre del 2024.
Sin duda, es un motivo de alegría cuando una empresa lanza un nuevo medicamento al mercado porque implica la culminación de un proyecto que en promedio dura entre 10 y 15 años y lleva aparejada una inversión de capital aproximada de 2,500 millones de euros, y por supuesto la obtención de las patentes involucradas. Estos proyectos incluyen diferentes hitos que involucran el desarrollo de la molécula, los estudios clínicos fase I, II y III, los planes de manejo de riesgo, la solicitud de autorización para comercialización a nivel global, la implementación de la cadena de suministro y el lanzamiento. Todo lo anterior, representa la oportunidad para curar o mejorar la calidad de vida de un paciente.

El lanzamiento de un nuevo medicamento toma entre 10 y 15 años e involucra una inversión de 2,500 millones de euros.
En estas líneas voy a referirme a los pacientes con cáncer. En este sentido, los nuevos desarrollos representan la oportunidad de lograr la remisión total de la enfermedad con la mayor mitigación posible del impacto colateral de la quimioterapia en el cuerpo, a fin de que, el paciente pueda continuar con su vida de la mejor forma posible. Cabe mencionar que, aunque las propuestas terapéuticas más modernas son muy eficaces, son muy caras; por lo que los tratamientos están disponibles, pero en la mayoría de los casos, los pacientes no pueden pagarlos de su bolsa y por eso se les denomina enfermedades catastróficas. Por este motivo los pacientes recurren a las coberturas de su seguro de gastos médicos mayores o a alguna institución pública de salud de la cual es derechohabiente. Cabe mencionar que ambos escenarios representan ventajas y desventajas; pero el paciente se ve en la necesidad de ajustarse a los protocolos y coberturas de las aseguradoras o de las instituciones públicas de salud. Incluso en algunos casos, los pacientes optan por los tratamientos en hospitales privados sin prever que hay coberturas y cláusulas para las recidivas; esta situación con frecuencia obliga al paciente a optar la continuidad del tratamiento a través de las instituciones de salud pública. Por otro lado, las instituciones públicas de salud siguen protocolos de tratamiento de primera línea que incluyen cirugía, radioterapia y conforme el caso lo requiera, la quimioterapia. Estos tratamientos pueden ser eficaces para algunos casos, pero en otros, se generan recidivas o procesos metastásicos que causan la muerte al 46% de los pacientes tratados. Generalmente los tratamientos con medicamento de patente se reservan a protocolos de segunda línea porque son muy caros y muy pocos pacientes tienen acceso a éstos. Ante esta situación, la opción del medicamento genérico puede significar la diferencia entre la vida y la muerte ya que, representa la oportunidad de que muchos más pacientes puedan recibir el tratamiento y por consiguiente maximizar la probabilidad de supervivencia. Pero en el balance del negocio, el vencimiento de una patente implica la reducción drástica de ingresos a la empresa innovadora, por lo que estas buscan extender la protección de la patente con nuevas indicaciones terapéuticas o el uso de nuevas tecnologías. Esto ocasiona por otro lado, que demore más tiempo la entrada del genérico o biosimilar al mercado, aún con la cláusula Bolar (excepción regulatoria), que permite realizar los estudios, pruebas y producción experimental correspondiente, dentro de los 3 años anteriores al vencimiento de la patente, para el caso de moléculas pequeñas. Para el caso de los biosimilares se permite la presentación anticipada del producto ante el Comité de Moléculas Nuevas (CMN) dentro de los 8 años anteriores al vencimiento de la patente. Por lo anterior, el desarrollo del medicamento genérico o biosimilar entraña desafíos enormes, al menos en México, ya que el Certificado de Buenas Prácticas de Fabricación de la planta de fabricación del Ingrediente Activo y del Producto Terminado deben ser otorgados por COFEPRIS o una agencia regulatoria reconocida por Ella. Además, los estudios de intercambiabilidad o biocomparabillidad deben realizarse en establecimientos aprobados por COFEPRIS o una Agencia Regulatoria reconocida y además con población Latina. Dado lo anterior, las propuestas de India, China, Bangladesh, Indonesia, entre otros, deben considerar en sus estrategias regulatorias la visita de verificación de COFEPRIS o de una Agencia Regulatoria de alta vigilancia. Adicionalmente, COFEPRIS, usualmente no acepta los estudios de intercambiabilidad o biocomparabilidad de dichos países por que no incluyen población latina y los criterios de aceptación están parcialmente alineados con las guías ICH. En tal caso, las empresas deben hacer una alineación con COFEPRIS para verificar si los protocolos clínicos existentes y los criterios de aceptación están alineados con la normatividad local y en su caso, realizar las alineaciones correspondientes para que la H. Autoridad apruebe los protocolos clínicos y puedan ejecutarse con población latina, lo que ocasiona que los proyectos se alarguen por varios años, en especial en el caso de los medicamentos biosimilares.
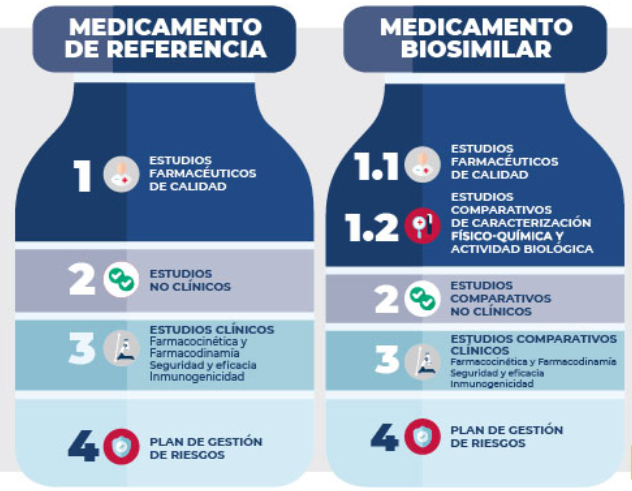
Las pruebas de intercambiabilidad y biocomparabilidad son necesarias para obtener la autorización para comercialización y se realizan para demostrar que cumplen con los criterios de aceptación establecidos en la NOM 177, al compararlos con el medicamento de referencia.
La obtención del registro sanitario es un hito muy importante porque el resultado no está en el total control de la empresa. El tiempo para obtener este documento cuyo propósito es autorizar la comercialización del producto en el mercado local, oscila entre 18 y 24 meses. Es muy importante considerar que una vez que se autoriza el registro sanitario, se procede a implementar la cadena de suministro lo cual puede tomar entre 6 y 12 en México. Los tiempos pueden ser mucho más largos si la cadena de suministro involucra uno o varios establecimientos en el extranjero. Este hito, es fundamental porque el producto ya debe estar en el canal para poder realizar su lanzamiento.
Conforme a lo anterior, para lograr el lanzamiento de un producto oncológico, genérico o biosimilar al mercado puede demorar entre 4 y 9 años, si se aprovecha la cláusula bolar desde el primer minuto.
Concluyo que, si la Autoridad Sanitaria y las empresas innovadoras trabajan con una buena estrategia regulatoria, buenas prácticas regulatorias y acciones de “reliance”, sería posible reducir los tiempos de lanzamiento de los medicamentos protegidos por patente y de la misma forma potencializar la innovación y así dar paso a nuevos desarrollos, así como, nuevas tecnologías. Por otro lado, sería posible reducir el tiempo para que un genérico o biosimilar entre al mercado, máxime que la tendencia de los últimos 10 años muestra un incremento de pacientes de la tercera edad y un incremento de casos de los siguientes tipos de cáncer: hígado, útero, próstata, riñón, tiroides y mama. En este sentido también es muy importante mantener un piso parejo para todos, los locales y los extranjeros, para no desincentivar la innovación y la inversión local. El proceso de “reliance” no debe implicar la omisión de la autorización para comercialización más que en los casos establecidos en el Art. 132 del RIS en donde la política sanitaria debe fundamentarse en el balance que debe existir entre la protección del mercado local y la maximización del uso de los recursos para lograr el mayor acceso posible de medicamentos para las mexicanas y mexicanos, sin detrimento del cumplimiento de la normatividad aplicable y sin perder de vista los costos necesarios para fabricar y distribuir los medicamentos localmente.
Si quieres saber más sobre estrategias regulatorias para el registro de medicamento genéricos y biosimilares, contáctanos por WhatsApp al 5539892647, a través de nuestro sitio web: www.pharmaclims.com.